Die neue Verordnung (EU) 2017/746 über In-vitro-Diagnostika
Die neue Verordnung - IVDR
Medizinprodukte zur in-vitro-Diagnostik (IVD) unterscheiden sich von anderen Medizinprodukten dadurch, dass sie nur zur Untersuchung von Proben aus dem menschlichen Körper bestimmt sind. Dieser Aspekt ist insbesondere aufgrund der geringeren Risiken ein so wichtiges Unterscheidungsmerkmal, dass IVD in einer eigenen Richtlinie (98/79/EG) neben der Medizinprodukterichtlinie 93/42/EWG, geregelt werden. Mit der neuen Verordnung über Medizinprodukte (MDR), die seit Mai 2021 gültig ist, wurde auch eine neue IVD-Verordnung in Kraft gesetzt.
Die neue In-vitro-Diagnostika Verordnung, kurz IVDR (in vitro diagnostic regulation), ist In Kraft seit dem 25.05.2017 und hat eine 5 jährige Übergangsfrist, bis sie ab dem 26.05.2022 gültig wird. Ausnahmen und Abweichungen von dieser Übergangsfrist sind in Artikel 113 Abs. 3 der IVDR geregelt.
Die IVDR ist, wie auch die MDR, eine Verordnung und keine Richtlinie. Das bedeutet sie wird ab dem 26.05.2022 verbindlich in allen EU-Mitgliedstaaten sein. Die bisherige Richtlinie hatte nur 24 Artikel, die IVDR hat nun über 113 Artikel verteilt auf 10 Kapitel, dem hinzu hat sie 15 Anhänge.
Was ist ein IVD?
Artikel 2 Abs. 2 der Verordnung:
„In-vitro-Diagnostikum“ bezeichnet ein Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibrator, Kontrollmaterial, Kit, Instrument, Apparat, Gerät, Software oder System — einzeln oder in Verbindung miteinander — vom Hersteller zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschließlich Blut- und Gewebespenden, bestimmt ist und ausschließlich oder hauptsächlich dazu dient, Informationen zu einem oder mehreren der folgenden Punkte zu liefern
a) über physiologische oder pathologische Prozesse oder Zustände,
b) über kongenitale körperliche oder geistige Beeinträchtigungen,
c) über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
d) zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
e) über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
f) zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Probenbehältnisse gelten als auch In-vitro-Diagnostika;“
Erweiterung der IVD Begriffsbestimmung
Die Beschreibung „kongenitale körperliche oder geistige Beeinträchtigungen“ ersetzt die Formulierung „angeborene Anomalien“ aus der Richtlinie.
Die Definition von IVD wird nun weiter gefasst. Die Definition unter c) über die Prädisposition, schließt auch prädiktive Gentests bzw. diagnostische Gentests ein.
Die Definitionen unter e) und f) sind Neuerungen, die im Bezug auf die personalisierte Medizin bzw. der therapiebegleitenden Diagnostika (Companion) aufgenommen wurden.

Point-of-Care Diagnostik
Produkte zur Eigenanwendung sind Produkte, die vom Hersteller zur Anwendung durch Laien bestimmt sind (z.B. Schwangerschaftstests). Daran hat sich nichts geändert.
Companion Diagnostics
„Therapiebegleitendes Diagnostikum“ bezeichnet ein Produkt, das für die sichere und wirksame Verwendung eines dazugehörigen Arzneimittels wesentlich ist, um
a) Patienten vor und/oder während der Behandlung zu identifizieren, die mit der größten Wahrscheinlichkeit von dem dazugehörigen Arzneimittel profitieren, oder
b) Patienten vor und/oder während der Behandlung zu identifizieren, bei denen wahrscheinlich ein erhöhtes Risiko von schwerwiegenden unerwünschten Reaktionen infolge einer Behandlung mit dem dazugehörigen Arzneimittel besteht; (IVDR Art. 2 Nr. 7).


Software
Die IVDR unterscheidet zwischen Software für IVD und Software für allgemeine Zwecke (Erwägunggrund 17 IVDR). Demnach ist Software dann ein IVD wenn sie gemäß der Definition für IVD, für medizinische Zwecke genutzt wird. Fehlt die medizinische Zweckbestimmung, handelt es sich um eine Software für allgemeine Zwecke, selbst wenn sie in Einrichtungen des Gesundheitswesens eingesetzt wird.
Witschaftsakteure
Klassifizierung von IVD
Die Änderung mit dem größten Einfluss auf die Industrie ist das neue System zur Klassifizierung von IVD. Dies stellt Hersteller und Benannte Stellen vor große Herausforderungen, bringt aber auch deutlich mehr Patientensicherheit.
In der IVD-Richtlinie 98/79/EG wurden eine Handvoll ausgewählter IVD mit höherer Risikoeinschätzung in Liste A und Liste B eingestuft. Diese Listen waren sehr kurz. Zu den Liste A Produkten zählten HIV- und Hepatitis-Tests sowie Reagenzien zur Blutgruppenbestimmung. Die Liste B war nur unbedeutend länger. Der gesamte Rest wurde zu den sog. „other Products“ bzw. „sonstigen Produkten“ gezählt.
Bisher konnten Hersteller für alle Produkte, die zu den „sonstigen Produkten“ zählten, ein selbst verantwortetes Konformitätsbewertungsverfahren durchführen und die Produkte mit einer Herstellerselbsterklärung (auch Eigenkonformitätserklärung), mit einem CE-Kennzeichen in Verkehr bringen. Ausschließlich für Liste A und Liste B Produkte musste die Benannte Stelle in der Konformitätsbewertung involviert werden bzw. für Liste A zusätzlich das Paul Ehrlich Institut (PEI) als überwachende Behörde bei der Chargenprüfung und -freigabe.
Dieses System konnte dem tatsächlichen Anspruch eines risikobasierten Klassifizierungssystems, im Sinne der Patientensicherheit, nicht gerecht werden.
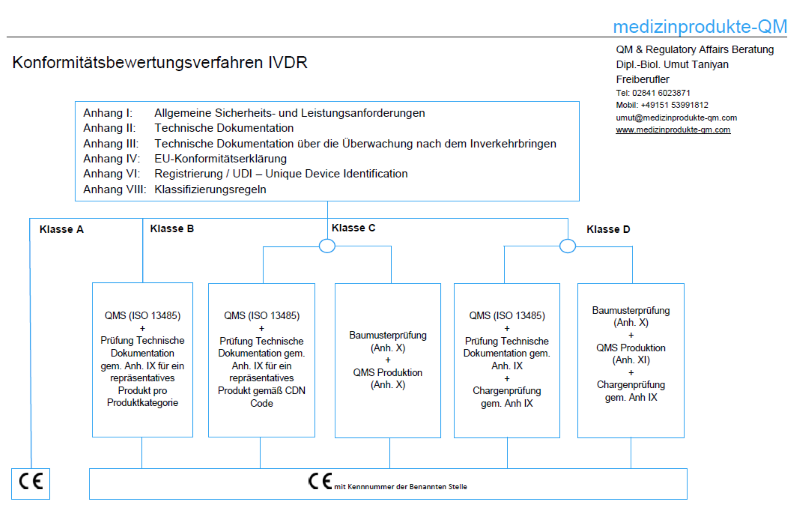
Mit der IVDR wird nun ein risikobasiertes Klassifizierungssystem mit Klassen von A bis D eingeführt. Die Klassifizierungsregeln werden in Anhang VIII beschrieben. Neben den Durchführungsvorschriften gibt es insgesamt 7 Klassifizierungsregeln. Jeder Hersteller ist selber dafür verantwortlich seine Produkte gemäß der Zweckbestimmung korrekt zu klassifizieren. Nachfolgend nur einige Beispiele und Auszüge. Es wird jedem Hersteller dringend empfohlen, sein Produktportfolio gemäß den Klassifizierungsregeln der IVDR zu prüfen.
Klasse A
Geringster Risikograd
Erzeugnisse für den allgemeinen Laborbedarf, Zubehör ohne kritische Merkmale, Pufferlösungen, Waschlösungen sowie allgemeine Nährmedien und histologische Färbungen…
Instrumente, die vom Hersteller speziell für die Verwendung bei In-vitro-Diagnoseverfahren vorgesehen sind;
Probenbehältnisse.
Klasse B
Mittlerer oder geringer Risikograd
Gemäß Regel 6: Produkte, die nicht unter die zuvor beschriebenen Klassifizierungsregeln fallen, werden der Klasse B zugeordnet.
Regel 7: Produkte bei denen es sich um Kontrollgeräte ohne einen zugewiesenen quantitativen oder qualitativen Wert handelt, werden der Klasse B zugeordnet.
Z.B. Schwangerschaftstest zur Eigenanwendung.

Klasse C
Hoher und Mittlerer Risikograd
Produkte, die zur Blutgruppenbestimmung oder Gewebetypisierung verwendet werden… (mit Ausnahme v. Produkten zur Bestimmung bestimmter Marker, s. Klasse D)
Produkte zur Eigenanwendung (z.B. Blutzuckermessgeräte zur Eigenanwendung), ausgenommen Schwangerschaftstests (Kl. B)
Produkte zum Einsatz zur Krebsvorsorge (z.B. PSA Tests).
Klasse D
Höchster Risikograd
Nachweis des Vorhandenseins von oder der Exposition ggü. übetragbaren Erregern im Blut…
…Erregern, die eine lebensbedrohende Krankheit mit einem hohen oder mutmaßlich hohen Verbreitungsrisiko verursachen;…
Z.B: HIV-Tests
Konformitätsbewertung
Das Konformitätsbewertungsverfahren ist vor dem Inverkehrbringen gemäß den Anhängen IX oder X & XI der IVDR durchzuführen. Je höher die Produktklasse ist, desto umfangreicher ist das Verfahren.
Konformitätsbewertungsverfahren für Produkte der Klasse A bleiben in der Verantwortung der Hersteller (Eigenkonformitätserklärung). Nach der bisherigen Richtlinie konnten 80% bis 90% aller IVD auf diese Weise in den Markt gebracht werden (sog. other IVD-products). Dies ändert sich nun radikal. Nach der IVDR werden nur noch ca. 10% bis 20% der IVD in Klasse A eingestuft werden*. Produkte der Klassen B, C, D erfordern die Mitwirkung von benannten Stellen (Notified Bodies), um das Konformitätsbewertungsverfahren durchzuführen.
Leistungsbewertung und klinischer Nachweis
Die Daten zur wissenschaftlichen Validität, zur Analyseleistung und zur klinischen Leistung, ihre Bewertung und der daraus abgeleitete klinische Nachweis werden in dem „Bericht über die Leistungsbewertung“ gemäß Anh. XIII Teil A Abschnitt 1.3.2 dokumentiert. Der Bericht wird der technischen Dokumentation beigefügt.
Der Bericht zur Leistungsbewertung und die dazugehörigen Unterlagen sind während des gesamten Lebenszyklus des Produkts zu aktualisieren. Die Daten zur Aktualisierung stammen aus der Durchführung des Plans für die Nachbeobachtung der Leistung nach dem Inverkehrbringen des Herstellers gemäß Anhang XIII Teil B und dem Plan zur Überwachung nach dem Inverkehrbringen gemäß Artikel 79.
Für Produkte der Klasse C und D ist der Bericht nach Bedarf, mindestens aber 1x jährlich zu aktualisieren.