Leistungen - In-vitro-Diagnostika
Als Biologe, Qualitätsmanager und Regulatory Affairs unterstütze ich Sie gezielt im Bereich der In-vitro-Diagnostik.
Durch meine langjährige Erfahrung mit diesen Produkten und Prozessen kenne ich nicht nur die regulatorischen Anforderungen der IVDR im Detail, sondern auch viele Spezifikationen und Testprinzipien von IVD – Unterstützung von der Entwicklung über die Herstellung bis hin zur Anwendung in der Praxis.
Ich begleite Sie kompetent durch alle Phasen – von der Klassifizierung über die Konformitätsbewertung bis zur CE-Kennzeichnung. Die Kommunikation mit Benannten Stellen und Aufsichtsbehörden gestalte ich für Sie auf Augenhöhe – klar, fachlich fundiert und lösungsorientiert.
So entlaste ich Ihr Team und trage dazu bei, dass Ihre Produkte effizient, regelkonform und praxisgerecht auf den Markt gelangen.
Die neue Verordnung - IVDR
In-vitro-Diagnostika (IVD) sind Medizinprodukte, die zur Untersuchung von Proben aus dem menschlichen Körper – etwa Blut oder Gewebe – bestimmt sind. Aufgrund ihres speziellen Anwendungsbereichs unterliegen sie einer eigenständigen regulatorischen Kontrolle innerhalb der Europäischen Union.
Die EU-Verordnung über In-vitro-Diagnostika (IVDR) ist seit dem 26. Mai 2022 verbindlich anzuwenden. Sie wurde am 25. Mai 2017 verabschiedet und ersetzt nach das bisherige Regelwerk.
Als unmittelbar geltendes EU-Recht bringt die IVDR umfangreiche Anforderungen mit sich: Über 113 Artikel in 10 Kapiteln sowie 15 Anhänge regeln die Sicherheit, Leistungsbewertung und Marktüberwachung von IVD-Produkten in der EU.
Was ist ein IVD?
„In-vitro-Diagnostikum“ bezeichnet ein Medizinprodukt, das als Reagenz, Reagenzprodukt, Kalibrator, Kontrollmaterial, Kit, Instrument, Apparat, Gerät, Software oder System — einzeln oder in Verbindung miteinander — vom Hersteller zur In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschließlich Blut- und Gewebespenden, bestimmt ist und ausschließlich oder hauptsächlich dazu dient, Informationen zu einem oder mehreren der folgenden Punkte zu liefern
a) über physiologische oder pathologische Prozesse oder Zustände,
b) über kongenitale körperliche oder geistige Beeinträchtigungen,
c) über die Prädisposition für einen bestimmten gesundheitlichen Zustand oder eine bestimmte Krankheit,
d) zur Feststellung der Unbedenklichkeit und Verträglichkeit bei den potenziellen Empfängern,
e) über die voraussichtliche Wirkung einer Behandlung oder die voraussichtlichen Reaktionen darauf oder
f) zur Festlegung oder Überwachung therapeutischer Maßnahmen.
Probenbehältnisse gelten als auch In-vitro-Diagnostika;“
Klassifizierung von IVD
Die Änderung mit dem größten Einfluss auf die Industrie ist das neue System zur Klassifizierung von IVD.
Dies stellt Hersteller und Benannte Stellen vor große Herausforderungen, bringt aber auch deutlich mehr Patientensicherheit mit sich.
In der IVD-Richtlinie 98/79/EG wurden eine Handvoll ausgewählter IVD mit höherer Risikoeinschätzung in Liste A und Liste B eingestuft. Diese Listen waren sehr kurz. Zu den Produkten der Liste A zählten HIV- und Hepatitis-Tests sowie Reagenzien zur Blutgruppenbestimmung. Die Liste B war nur geringfügig länger. Der gesamte übrige Teil wurde den sogenannten „sonstigen Produkten“ (engl. „other products“) zugeordnet.
Bisher konnten Hersteller für alle Produkte, die zu den „sonstigen Produkten“ gehörten, ein eigenverantwortliches Konformitätsbewertungsverfahren durchführen und diese mittels einer Herstellerselbsterklärung (auch Eigenkonformitätserklärung) mit CE-Kennzeichnung in Verkehr bringen. Ausschließlich für Produkte der Liste A und Liste B musste eine Benannte Stelle in das Konformitätsbewertungsverfahren einbezogen werden – bei Produkten der Liste A zudem das Paul-Ehrlich-Institut (PEI) als überwachende Behörde bei der Chargenprüfung und -freigabe.
Dieses System wurde dem tatsächlichen Anspruch eines risikobasierten Klassifizierungssystems im Sinne der Patientensicherheit nicht gerecht.
Mit der IVDR wird nun ein risikobasiertes Klassifizierungssystem mit den Klassen A bis D eingeführt. Die Klassifizierungsregeln sind in Anhang VIII beschrieben. Es gibt insgesamt sieben Klassifizierungsregeln, ergänzt durch Durchführungsvorschriften. Jeder Hersteller ist selbst dafür verantwortlich, seine Produkte entsprechend der Zweckbestimmung korrekt zu klassifizieren.
Nachfolgend einige Beispiele und Auszüge. Es wird jedem Hersteller dringend empfohlen, das gesamte Produktportfolio anhand der Klassifizierungsregeln der IVDR zu überprüfen.

Klasse A
Geringster Risikograd
Erzeugnisse für den allgemeinen Laborbedarf, Zubehör ohne kritische Merkmale, Pufferlösungen, Waschlösungen sowie allgemeine Nährmedien und histologische Färbungen…
Instrumente, die vom Hersteller speziell für die Verwendung bei In-vitro-Diagnoseverfahren vorgesehen sind;
Probenbehältnisse.
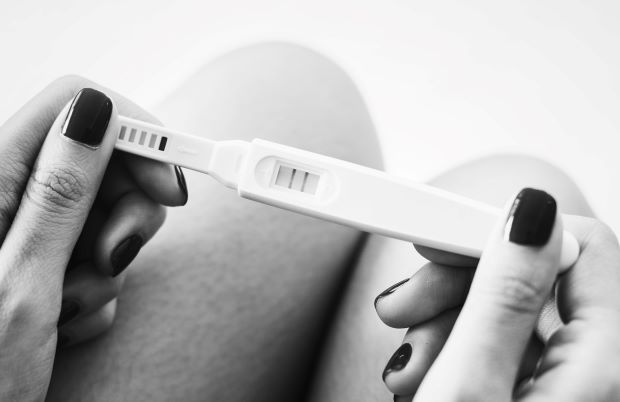
Klasse B
Mittlerer oder geringer Risikograd
Gemäß Regel 6: Produkte, die nicht unter die zuvor beschriebenen Klassifizierungsregeln fallen, werden der Klasse B zugeordnet.
Regel 7: Produkte bei denen es sich um Kontrollgeräte ohne einen zugewiesenen quantitativen oder qualitativen Wert handelt, werden der Klasse B zugeordnet.
Z.B. Schwangerschaftstest zur Eigenanwendung.